联系我们
检测服务:400-803-7959
其他咨询:400-650-9231

检测服务:400-803-7959
其他咨询:400-650-9231
| 基因与疾病 |儿童心肌病的分类及诊断之肥厚型心肌病 |
| 时间:2019-06-17 16:28:47 |
|
近日,Circulation发表了一篇来自美国心脏协会的科学声明,在该声明中儿童心肌病领域的专家就儿童心肌病病因及诊断进行了阐述,期望这份声明推动儿童心肌病早诊断、早治疗、提高患儿生活质量。 儿童心肌病发病率低,为1/100,000,但是近40%有症状的心肌病儿童接受了心脏移植或在确诊后两年内死亡。美国国家心肺血液研究所资助的儿童心肌病登记信息表明,只有很少一部分心肌病患儿能确定病因,大多数患儿可能存在遗传因素。 肥厚型心肌病是儿童心肌病的常见原因,占儿童和少年心肌病的30-50%。肥厚型心肌病患病率在小于1岁的孩子中尤其高,是大于1岁孩子的3倍。下面详细介绍美国心脏协会科学声明中关于儿童肥厚型心肌病的病因及诊断方法。
肥厚型心肌病 ◆ 定义 肥厚型心肌病(HCM)是一种以心肌肥厚为特征的心肌疾病,主要表现为室间隔或左心室壁增厚,通常不伴有左心室腔的扩大,需排除生理性肥大(即继发于体力活动)和病理性肥大(即继发于高血压、主动脉瓣狭窄和其他疾病)。 ◆ 诊断标准 HCM的主要诊断标准是室间隔或左室壁厚度。成人诊断标准是室间隔或左室壁厚度≥15mm,或者有明确家族史者成人≥13mm。成人的标准不根据身体尺寸进行调整,这就造成了诊断不足或诊断过度的风险。 儿童必须根据身体尺寸进行调整,因此,诊断标准是基于壁厚Z评分,根据同体表面积正常儿童左室壁厚平均值加SD计算。儿童的特定Z评分阈值尚未验证,因此建议采用成人壁厚标准的Z评分。 儿童HCM的病因 儿童HCM病因分为:肌小节蛋白基因突变、综合征、糖原和脂肪酸代谢异常、溶酶体贮积症、线粒体病和母亲患有糖尿病(Table 7)。下面将分别讨论这些病因。
◆ 肌小节蛋白基因突变性HCM 存在肌小节蛋白基因致病性变异或可能致病性变异的同时符合HCM表型诊断标准为肌小节蛋白基因突变性HCM。家族性HCM也被认为是肌小节蛋白基因突变性HCM,呈典型的常染色体显性遗传。目前,40%-50%符合表型诊断标准的个体基因检测结果阴性,因此非肌小节蛋白基因突变也是可能的病因。基因检测阴性个体的治疗与有明确基因突变个体的治疗大体相同,因此通常将它们组合在一起。 肌小节蛋白相关基因最初只包含8个 (MYH7、 MYL2、 MYL3、 MYBPC3、 TNNT2、 TNNI3、TPM1、 ACTC1),后来发现了与肌小节蛋白基因突变有相同表型的其它基因突变,包括Z-盘蛋白(如:CSRP3、ACTN2)和钙调节蛋白(如:PLN)。尽管有大量的基因和突变被认为引起肌小节蛋白突变性HCM,但其分子机制还不能确定。功能获得性突变被认为是引起疾病的主要原因。 在同一家族中,肌小节蛋白基因突变既与DCM相关又与HCM相关,其机制是导致肌张力降低的突变引起DCM,导致肌张力升高的突变引起HCM。 肌小节蛋白基因突变性HCM的诊断标准依赖于存在肌小节蛋白基因突变同时具有HCM表型。如果没有基因检测证实肌小节基因突变,具备一定的肌小节基因突变性HCM表型特征通常也被假设是肌小节蛋白基因突变性HCM。 ◆ 继发性的HCM 1. 糖原代谢异常 糖原贮积病(GSDs)是糖原储存或使用异常的先天性代谢障碍疾病。最常见引起HCM表型的疾病包括GSD II型(Pompe病)、Danon病和PRKAG2心肌病。糖原贮存病III型也可伴有HCM。 Pompe病是α-1,4葡糖苷酶活性缺陷引起的常染色体隐性遗传病。婴儿型、少年型和成人型已被描述,其中婴儿型最可能表现为早期严重的心肌肥厚。如果婴儿出现巨大的心脏肥大,特征性的心电图(短PR间期,高大的QRS波),并伴有低肌张力,应怀疑Pompe病。通过GAA酶活性和基因检测进行确诊。 Danon病是一种由LAMP2基因缺陷引起的X连锁显性遗传病。Danon病最突出的表现是HCM、骨骼肌异常和轻度智力障碍。男性患者往往比女性患者更早出现严重的心脏受累,鉴于该病进展相对较快,且难治性心律失常频繁发生,应尽早通过基因检测确诊。 PRKAG2基因的突变导致糖原积累和心脏肥大,以及电生理异常。任何出现典型的HCM三联征、心室预激和进行性传导障碍的病人都应怀疑为PRKAG2型心肌病。应通过基因检测确诊。 2. 溶酶体贮积症 溶酶体贮积病是一类异质性遗传病,其特征是机体生物大分子不能被正常降解,导致细胞功能障碍和器官肿大。最常见的引起HCM的形式包括粘多糖病、粘脂病和鞘脂病。 粘多糖贮积症I型(Hurler综合征)是一种常染色体隐性遗传病,而粘多糖贮积症II型(Hunter综合征)是一种X连锁隐性遗传病。两者的临床表现都是身材矮小、面容粗陋、骨骼异常和HCM。可能比HCM更常见的心脏异常是瓣膜组织中糖胺聚糖沉积引起的瓣膜异常。诊断依据是尿液中糖胺聚糖浓度升高,并经基因检测确诊。 粘脂质贮积症II型是一种常染色体隐性遗传病,该病是由于N-乙酰氨基葡萄糖-1-磷酸转移酶缺乏,导致酸性水解酶分泌到血浆中,而不与溶酶体结合所致。该病的组织学特征是存在I细胞,它是带有包涵体的成纤维细胞。除HCM外,其他临床特征包括面容粗陋、皮肤增厚、骨骼异常和瓣膜异常。诊断依赖于尿寡糖和血浆溶酶体水解酶浓度,以及GNPTAB基因检测。 法布雷病是由α-半乳糖苷酶A缺乏引起的X连锁隐性遗传病。酰基鞘氨醇三己糖苷在各种组织和器官的溶酶体中的积累导致包括神经系统、肾功能不全和胃肠道疾病的表现。这种多器官疾病的症状和体征发生在青少年期,通常为血管角化瘤和胃肠道疼痛。心脏受累包括向心性肥大,常发生于生命的第40年和第50年。然而,由于该病发病早,即使在儿童身上也能检测到细微的心脏异常。进行性心室肥大是该病的典型心脏表现,导致舒张功能障碍和心力衰竭,最初收缩功能保持不变,随后心室扩张伴收缩功能障碍。瓣膜受累较少见,但可导致瓣膜功能不全或狭窄。高血压可发生于肾功能不全的成年人。通过检测α-半乳糖苷酶A的酶活性,可以确诊男性患者。GLA基因突变检测对女性患者的确诊是必要的。 3. 综合征 新生儿期或婴儿期的HCM常出现在一些临床综合征(如:努南综合征及其类似疾病)及心血管异常疾病(如肺动脉瓣狭窄、二尖瓣发育不良、房间隔缺损和室间隔缺损)。努南综合征是一种常染色体显性遗传病,表现为身材矮小、发育迟缓、眼距宽、颈蹼、下斜睑裂和上睑下垂。当存在表型特征时,基因检测可辅助诊断。努南综合征中HCM的患病率为22%,其中约半数患者在6个月前确诊,远远早于非综合征性HCM患儿。与非综合征HCM患儿相比,努南综合征HCM患儿更容易出现充血性心力衰竭(24% VS 9%),左心室流出道阻塞(30% VS 9%),3年死亡率更高(26% VS 11%)。HCM在Costello综合征中也很常见,超过一半的确诊患者都有HCM。与努南综合征相似,非对称性房间隔肥大是其典型的特征,左室流出道阻塞也可能发生。Noonan和Costello综合征与心面皮肤综合征有表型重叠。这种疾病很少发生,但是HCM患者中发生率很高(44%) 。发生HCM的其它综合征包括Donohue综合征、Sawyer综合征和BeckwithWiedemann综合征。与这些综合征相关的许多基因已被确定(Table 8)。
4. FriedReich共济失调 Friedreich共济失调是一种常染色体隐性遗传病,最常见的病因是FXN基因中的GAA三核苷酸重复扩增。这种扩张导致了Frataxin的缺乏,Frataxin是一种参与线粒体和细胞铁稳态的蛋白质。患有进行性共济失调、无条件反射和HCM的儿童应怀疑Friedreich共济失调。心律失常和DCM有时可见,更常见于成年期。 5. 脂肪酸代谢异常 脂肪酸代谢异常许多情况下都可出现DCM或HCM。有一种情况似乎主要与HCM有关,那就是长链酰基辅酶A脱氢酶缺乏。其他表现为HCM疾病通常包括多种酰基辅酶A脱氢酶、长链羟基辅酶A脱氢酶、脂酰肉碱转位酶、肉碱棕榈酰转移酶II和肉碱-脂酰肉碱转位酶缺乏症。 6. 高胰岛素血症 由胰岛素过量引起的心肌病只发生在新生儿中。高胰岛素血症可分为原发性和继发性,无论哪种形式,它都被认为是先天性心肌病。 在原发性高胰岛素症中,胰腺的β胰岛细胞可发生局灶性腺瘤转化或弥漫性功能亢进,两者都会导致胰岛素的过度分泌。婴儿表现为严重低血糖症、癫痫、低温、低肌张力和嗜睡。在一项研究中,有68名<3个月的婴儿患有这种疾病,其中25名发现呼吸窘迫、胸片异常、杂音或心律失常,在这25例中,有10例发现有HCM表现,所有患者均接受高胰岛素血症治疗,HCM均消失。 在继发性高胰岛素症中,婴儿没有胰腺组织学异常。这类婴儿被认为是由于母亲糖尿病或妊娠期糖尿病引起胎儿产生过多胰岛素。认识到母亲糖尿病和监测血糖的重要性,将有助于管理新生儿。尽管这些婴儿可能出现心力衰竭,但HCM在出生后的第一年就消退了。 7. 肢端肥大症 肢端肥大症是由于垂体腺瘤分泌过多生长激素而导致的一种罕见疾病。糖代谢紊乱伴高胰岛素和葡萄糖耐受不良是包括心血管系统在内的多系统异常的基础。临床表现包括头、手、脚、胸和心脏肿大。在生长激素长期过量的情况下,可发生高血压、冠状动脉疾病和心肌肥厚。即使在较短的时间内过量,也会发展成左室肥大。当内分泌疾病得到治疗时,这些变化可逆转。 HCM的临床结局 HCM患儿的5年生存率(无死亡或移植)差异很大,从先天性代谢异常的HCM患儿的42%到1岁后出现HCM(非婴儿型特发性HCM)患儿的94% (Table 9)。
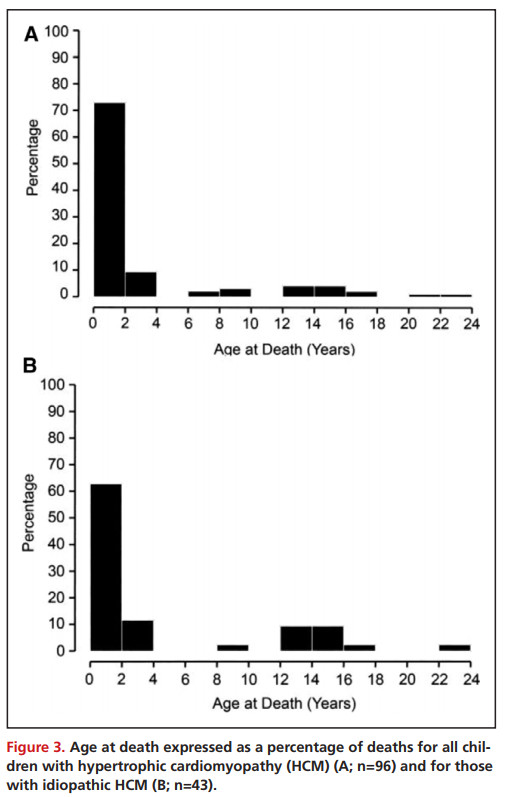
所有儿童HCM以及特发性HCM患者的死亡年龄分布均为双峰型,诊断后前2年死亡率最高,青春期死亡率峰值较小(Figure 3)。
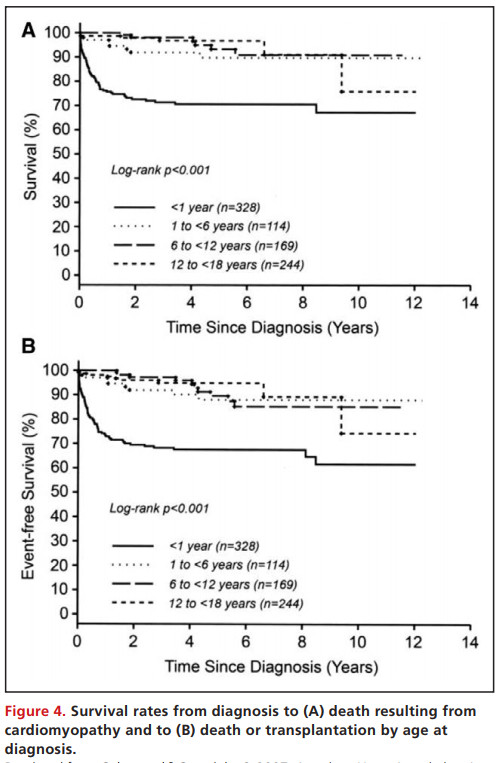
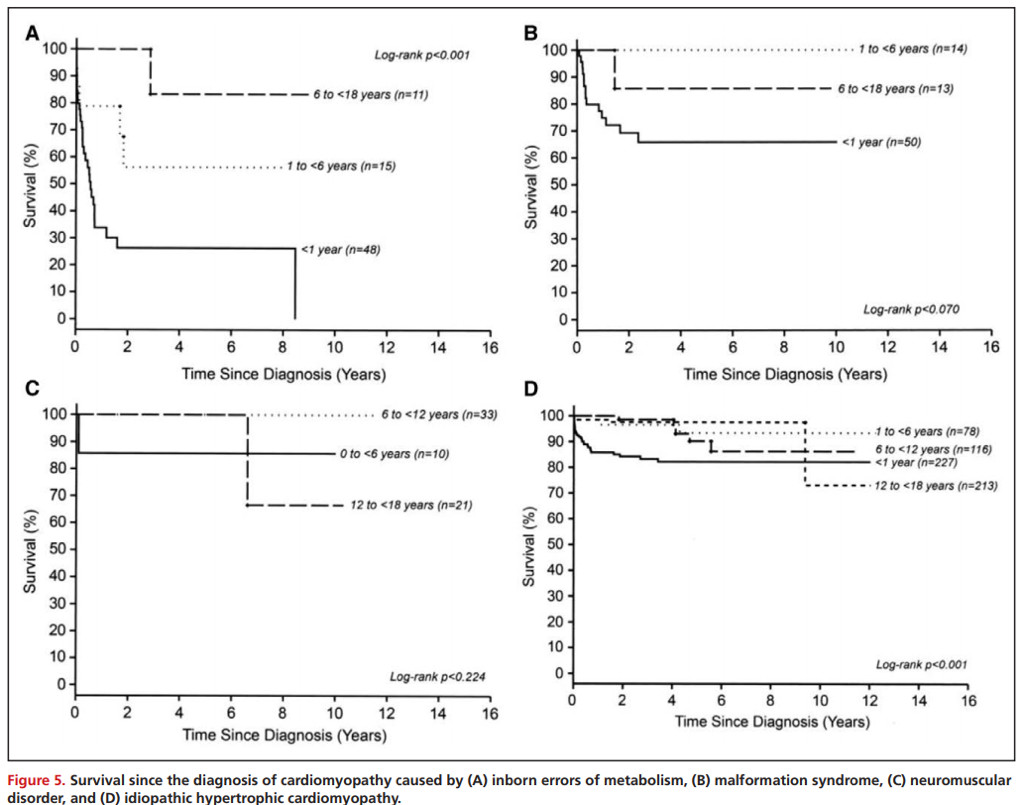
1岁以前出现HCM通常预后更差,特别是在患有特发性疾病的儿童和那些出现先天性代谢障碍或畸形综合征的儿童中(Figure 4和Figure 5)
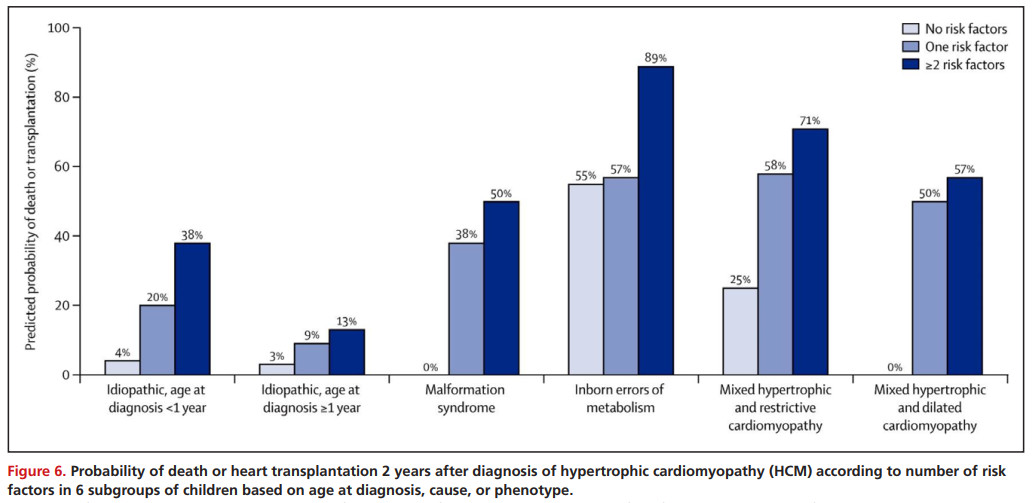
除了诊断时的年龄,心衰和表现较低的心肌缩短分数通常与不良结果有关。例如,小于6个月的努南综合征患儿在出现充血性心力衰竭时,Z分数低于人口中位数的儿童预后较差,近20%的死亡发生在生命的第一年。具有HCM表型且诊断时确定有2个以上危险因素的患儿预后较差,尽管不同的致病组的预后不同(Figure6)。对于儿童特发性HCM, 1岁后存活者的死亡率约为1 / 100,与成人的死亡率一致。
儿童肥厚型心肌病多由遗传因素导致,进行基因检测可以早期发现病因,指导正确治疗及家系管理,提高患者生活质量。 参考文献: Lipshultz SE,et al.Cardiomyopathy in Children: Classification and Diagnosis.Circulation. 2019. END
如果您想了解更多资讯或订购我们的服务,请您通过以下方式联系我们。 您也可以关注本公众号留下您的联系方式,我们会及时给您回复。 官网丨http://bestnovo.com 电话丨400-650-9231 关于百世诺 百世诺医疗提供一站式的心脑血管疾病整体解决方案,提供基因检测、遗传咨询、专家报告解读等服务,百世诺(北京)医疗科技有限公司是中国心脑血管精准医疗的领创者,总部位于北京市中关村生命科学园北大医疗产业园。公司下设百世诺(北京)医学检验实验室、江苏百世诺医疗科技有限公司、北京熙睿诊所三大分支机构。 公司的研发团队由北大、北医、协和、中科院等权威研究机构及医院的30余位心脑血管医学专家、医学遗传学家、生物信息学家及基因检测技术专家组成,拥有智能化生物信息分析软件和万人中国人群心血管病基因数据库,并不断优化、持续更新。
|